HuBMAP Liquid Chromatography Mass Spectrometry (LCMS)
Last Updated 6/22/2020
Overview
This document details mass spectrometry (mass spec, MS) assays, data states, metadata fields, file structure, QA/QC thresholds, and data processing.
Description
Coupling of liquid chromatography (LC) to mass spectrometry (MS) has become an indispensable technique for analyzing complex mixtures of biomolecules. Chromatography is a technique for separation of molecules based on their interactions with a stationary phase and a mobile phase of flowing solvent. For example, in reversed-phase chromatography, the stationary phase is commonly composed of hydrophobic C18-functionalized silica particles that are packed into a column, and the mobile phase is composed of a combination of water and acetonitrile. Based on their differing hydrophobicities, biomolecules partition differently between the stationary and mobile phases. Consequently, biomolecules elute off of a column at different times. Mass spectrometry measures the molecular weights of eluting biomolecules via detection of gas-phase ions. To obtain the masses of eluting biomolecules via mass spectrometry, solution-phase biomolecules are most commonly converted to gas-phase ions via electrospray ionization in which a high voltage is applied to the liquid coming out of the column. The following are descriptions of the various LC-MS assays that are being utilized for HuBMAP (Figure 1).
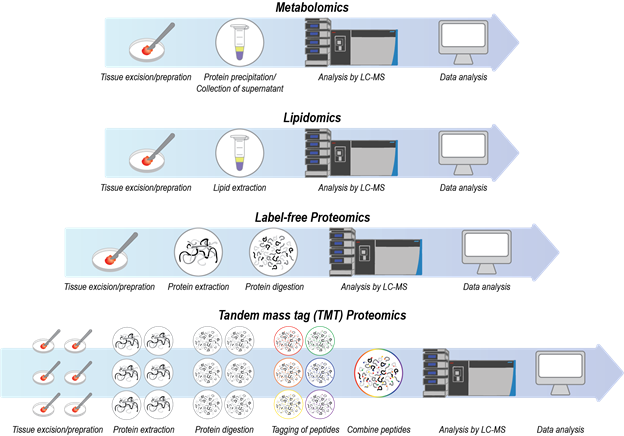 Figure 1. A visual depiction of the general sample preparation procedures for LC-MS assays.
Figure 1. A visual depiction of the general sample preparation procedures for LC-MS assays.
HuBMAP Data States (Levels)
The HuBMAP project provides data to the public in a variety of data states, which denote the amount of processing that has been done to the data. The data states for RNA seq data provided by the HuBMAP project are listed below:
| Data State | Description | LC/MS File Format |
|---|---|---|
| 0 | Raw data: raw MS data from instrument. | Raw, wiff |
| 1 | Txt, tsv, csv: MS intensity (spectral count) + Annotation | txt, tsv, csv |
HuBMAP Metadata
Definition of Metadata Levels
-
Level 1: These are attributes that are common to all assays, for example, the type (“CODEX”) and category of assay (“imaging”), a timestamp, and the name of the person who executed the assay.
-
Level 2: These are attributes that are common to a category of HuBMAP assays, i.e. imaging, sequencing, or mass spectrometry. For example, for imaging assays this includes fields such as x resolution and y resolution.
-
Level 3: These are attributes that are specific to the type of assay, for example for CODEX that would include number of antibodies and number of cycles.
-
Level 4: This is information that might be unique to a lab or is not required for reproducibility or is otherwise not relevant for outside groups. This information is submitted in the form of a single file, a ZIP archive containing multiple files, or a directory of files. There is no formatting requirement (although formats readable with common tools such as text editors are preferable over proprietary binary formats).
## Associated Files All HuBMAP data will have searchable metadata fields.
The metadata schema is available in Github for download.
HuBMAP QA/QC of raw (state0) data files
Untargeted Label-free proteomics utilizing MaxQuant
Untargeted data independent analysis (DIA) methods are assessed using a quality control (QC) pipeline for proteomics results generated by MaxQuant (PTXQC - https://github.com/cbielow/PTXQC). This is an open source software package that creates QC/QA reports with metrics and automated scoring functions. The automated scores are collated to create an overview heatmap as the first figure of the report (XML and PDF) to allow ease of interpretation, even by nonspecialists. A full outline of the metrics is highlighted below. These metrics address all four aspects of the experiment including sample preparation, LC, MS and general performance. These reports are provided for all LC-MS DIA data generated by the VU TMC.
 Outline of the full ProTeomiX (PTX) Quality Control (QC) Report including metrics provided.
Outline of the full ProTeomiX (PTX) Quality Control (QC) Report including metrics provided.
Overview
- 1.1 HeatMap
- 1.2 Name Mapping
- 1.3 Metrics
- 1.3.1 PAR: parameters
- 1.3.2 EVD: Top5 Contaminants per Raw file
- 1.3.3 EVD: Contaminants
- 1.3.4 EVD: peptide intensity distribution
- 1.3.5 PG: intensity distribution
- 1.3.6 MSMS: Missed cleavages per Raw file
- 1.3.7 EVD: charge distribution
- 1.3.8 PG: Contaminant per condition
- 1.3.9 MSMSscans: TopN over RT
- 1.3.10 EVD: IDs over RT
- 1.3.11 EVD: Peak width over RT
- 1.3.12 MSMSscans: Ion Injection Time over RT
- 1.3.13 [experimental] MSMSscans: MS/MS intensity
- 1.3.14 EVD: Oversampling (MS/MS counts per 3D-peak)
- 1.3.15 EVD: Uncalibrated mass error
- 1.3.16 EVD: Calibrated mass error
- 1.3.17 MSMS: Fragment mass errors per Raw file
- 1.3.18 SM: MS/MS identified per Raw file
- 1.3.19 MSMSscans: TopN
- 1.3.20 MSMSscans: TopN % identified over N
- 1.3.21 Missing Values Skipped
- 1.3.22 EVD: Peptide ID count
- 1.3.23 EVD: ProteinGroups count
See J. Proteome Res., 2016, 15 (3), pp 777-787. DOI: 10.1021/acs.jproteome.5b00780 for further details.
QC for Lipidomics & Metabolomics
In order to test for batch effects, PCA plots are generated to confirm correct clustering of quality-control-samples.
Terms defined in this document
| Term | Definition |
|---|---|
| TMT proteomics | To continuously monitor LC and mass analyzer performance, Hela cell tryptic digest was used as a quality control (QC) sample in this protocol. Proteins were extracted in 8M Urea lysis buffer from the Hela cell, then reduced and alkylated with DTT and IAA. After trypsin digestion overnight, peptides were cleaned up and labeled with TMTzero. The labeled peptides were analyzed by the same LC method without fractionation and MS3 instrument method. Chromatograph profile, mass accuracy, number of protein identification and TMT reporter ion intensity for quantitation were monitored to evaluate instruments stability and sensitivity. |
| Lipidomics | After each cleaning (front plate and DMS), more than 24 hours of idling or 3 days of consecutive use, the DMS compensation voltage is tuned using a set of lipid standards (cat# 945 5040141, Sciex). Before each batch, a quick system suitability test (QSST) (cat# 50407, Sciex) is performed to ensure an acceptable limit of detection for each lipid class. A maximum of 46 samples are processed and analyzed per batch. In addition, each batch contains a preparation blank as well as three lipid extracts from a reference plasma sample (QC, cat# 948 4386703, Sciex). Data quality is assessed by counting the number of detected lipid species in each sample (typically >800 lipid species). Also, QC samples should be clustered together as well as samples prepared and analyzed in different batches should be overlaid on a principal component analysis plot. |
| Metabolomics | After each cleaning (front plate, transfer capillary) that happens approximately every 4 days, mass calibration in positive and negative modes is performed using calibration mixes (cat# 88323, 88324, Thermo). Every 3 months, a more extensive calibration protocol including electronics, ion transfer, quadrupole and orbitrap is performed. Each sample is spiked-in with 15 labeled internal standards that are used to check mass accuracy, retention time and peak shape. Before each batch, the LC-MS system is equilibrated by injecting 6 and 12 pool samples (QC) for RPLC and HILIC, respectively. Signal deviation over time is controlled for by injecting a pool sample every 10 injections. After data processing, QC samples should be clustered together as well as samples prepared and analyzed in different batches should be overlaid on a principal component analysis plot. |
For Additional Help
- For general information on LC-MS, contact: Jeffrey Spraggins
- For questions about shotgun LC-MS (targeted lipidomics), contact: Kevin Contrepois
- For questions about TMT LC-MS (proteomics), contact: Lihua Jiang
- For questions about untargeted LC-MS/MS (untargeted proteomics), contact: Jeffrey Spraggins
- For questions about untargeted LC-MS (untargeted metabolomics), contact: Kevin Contrepois